
Les scientifiques de l’Hospital for Sick Children (SickKids) ont révélé une couche de variation génétique jusqu’alors négligée qui pourrait aider à expliquer pourquoi les gens vivent différemment la maladie et pourquoi certains traitements fonctionnent mieux pour certaines populations.
Les répétitions en tandem sont des sections répétées d’un brin d’ADN qui représentent environ sept pour cent du génome humain. La probabilité que ces répétitions en tandem provoquent des erreurs dans la fonction des gènes augmente à chaque fois qu’elles se répètent. On sait qu’elles provoquent des affections telles que la maladie de Huntington et sont impliquées dans de nombreuses autres, notamment les troubles du spectre autistique, la schizophrénie et la cardiomyopathie.
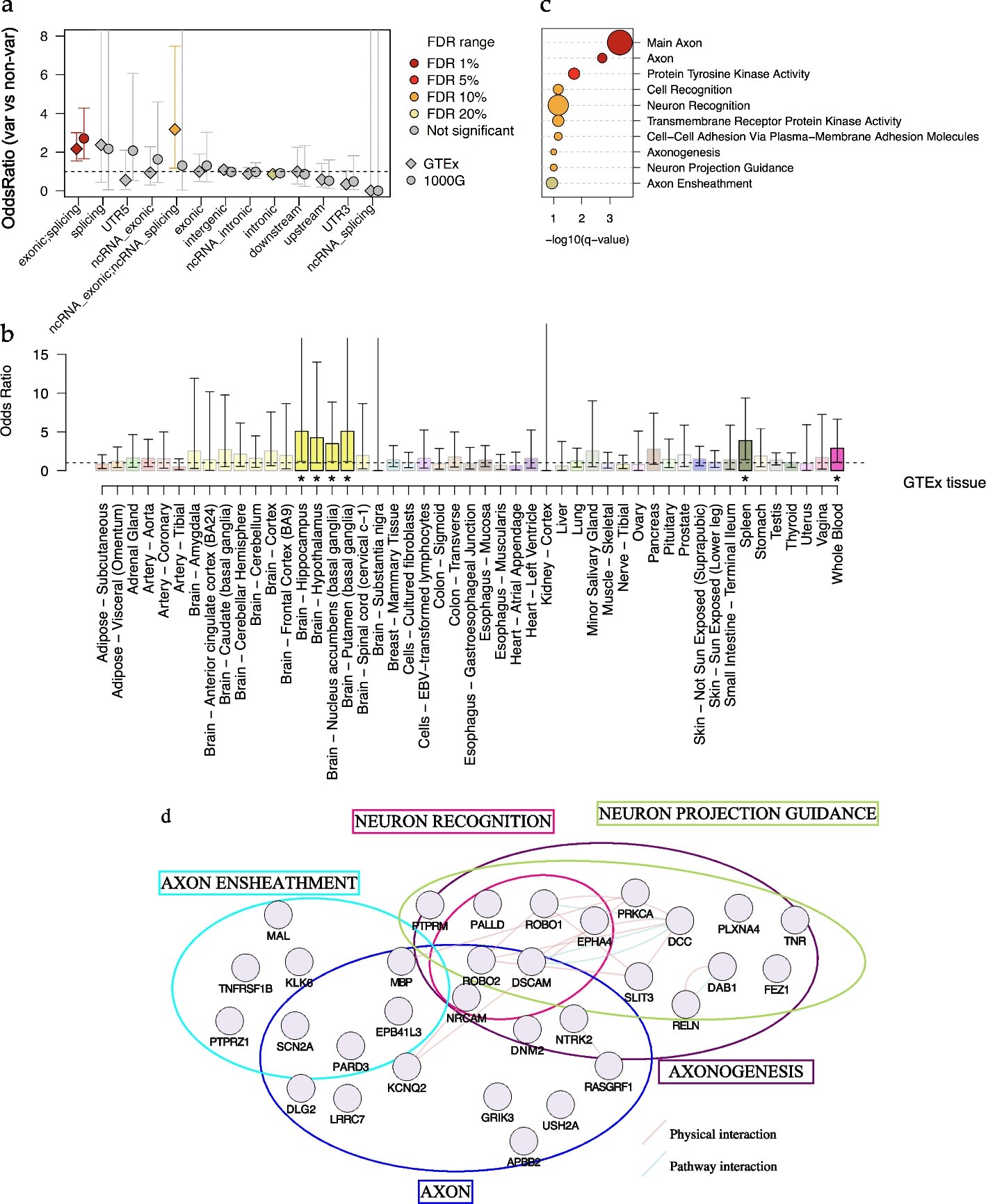
Aujourd’hui, une étude menée par SickKids est la première à montrer qu’en plus de la longueur de la répétition en tandem, des changements subtils dans la composition des courtes répétitions en tandem (STR) peuvent également avoir un impact important sur le fonctionnement des gènes.
Publié dans Biologie du génomel’équipe de recherche a analysé les données génomiques de plus de 3 000 individus de la population générale et a découvert qu’environ sept pour cent des STR du génome humain présentent des variations dans la composition des séquences.
« Ces changements dans la composition des STR ne sont pas rares, ils font partie intégrante de la diversité génétique humaine. Il s’agit d’une nouvelle dimension de variation génétique qui se cache à la vue de tous », explique le Dr Ryan Yuen, responsable de l’étude et scientifique principal du programme de génétique et de biologie du génome.
STR étroitement liés au développement et au fonctionnement du cerveau
En plus de décrire la prévalence des STR dans la population générale, l’équipe de recherche a fait plusieurs autres découvertes qui pourraient éclairer de futures recherches et développements thérapeutiques de précision à l’appui de Precision Child Health, un mouvement chez SickKids visant à individualiser les soins pour chaque patient.
Premièrement, ils ont découvert que les STR variables sont souvent situés à proximité d’éléments Alu, des séquences d’ADN répétitives dont la fonction est encore relativement inconnue. Ces STR apparaissent plus fréquemment aux jonctions d’épissage des gènes impliqués dans le développement et le fonctionnement du cerveau.
« Nous avons observé des tendances claires, comme ces diverses répétitions apparaissant dans les gènes liés au développement neurologique et au fonctionnement cérébral », explique le Dr Sasha Mitina, chercheur au Yuen Lab et premier auteur de l’étude. « Les gènes affectés par ces variations sont liés à des processus biologiques critiques et peuvent aider à expliquer les différences individuelles en matière de santé et de maladie. »
L’étude a également révélé des tendances distinctes dans la variabilité du STR parmi différents groupes ethniques, une découverte qui, selon l’équipe de recherche, pourrait aider les scientifiques à mieux prédire le risque de maladie et à adapter les traitements à différentes populations à l’avenir.
Une nouvelle dimension de la variation génétique
La plupart des outils et algorithmes ne prennent en compte que la longueur des répétitions en tandem. Utilisant un algorithme développé par Yuen et grâce à l’expertise du Centre de génomique appliquée (TCAG) de SickKids, l’équipe a pu détecter à la fois la taille et la composition des répétitions, même avec des données de séquençage à lecture courte.
« Notre approche nous permet de voir à la fois la taille et la composition des séquences », explique Yuen. « Nous ne faisons encore qu’effleurer la surface, mais ces régions pourraient détenir les réponses à certaines des inconnues de notre génome et contenir des cibles potentielles pour de futures études sur les maladies. »
À mesure que le séquençage à lecture longue devient plus courant, les chercheurs espèrent découvrir des variations encore plus cachées ainsi que de nouvelles connaissances sur les conditions neurodéveloppementales, éclairant ainsi l’avenir de la génomique clinique.