
Les scientifiques de l’hôpital de recherche pour enfants St. Jude et de l’université Northwestern ont identifié une opportunité de traitement jusqu’alors inconnue pour la drépanocytose et la β-thalassémie. La découverte, publiée dans Sangrepose sur une nouvelle compréhension du fonctionnement de la thérapie génique basée sur CRISPR.
Dans ces thérapies, CRISPR-Cas9 cible un élément régulateur de l’ADN, appelé amplificateur, qui contrôle l’expression de BCL11A, un gène clé responsable du passage de la production d’hémoglobine des formes fœtales aux formes adultes. Les chercheurs ont découvert que cela perturbait la structure tridimensionnelle du génome nécessaire au maintien d’un niveau élevé d’expression de BCL11A dans les précurseurs des globules rouges.
En conséquence, BCL11A est réduit au silence, conduisant à la réactivation de l’hémoglobine fœtale, qui compense l’hémoglobine adulte défectueuse dans la drépanocytose et la β-thalassémie. Les chercheurs ont en outre découvert que le ciblage d’un ARN spécifique produit par l’amplificateur BCL11A pourrait avoir des effets similaires à ceux de la thérapie génique.
BCL11A réprime l’expression de l’hémoglobine fœtale, qui est généralement produite à de faibles niveaux dans les globules rouges des adultes en bonne santé. L’inactivation de BCL11A peut réactiver l’expression de l’hémoglobine fœtale, compensant ainsi l’hémoglobine falciforme mutante dans la drépanocytose ou la perte des gènes de la β-globine dans la β-thalassémie.
Les thérapies géniques récemment approuvées pour ces troubles utilisent l’édition du génome basée sur CRISPR pour cibler l’amplificateur BCL11A dans les cellules souches sanguines, obtenant ainsi des résultats transformateurs pour les patients. Une compréhension plus détaillée de la façon dont la thérapie génique agit pour perturber l’expression de BCL11A reste cependant une question en cours.
Les coûts élevés, la disponibilité limitée et les risques potentiels associés aux thérapies géniques actuelles les rendent inaccessibles à la plupart des patients. Il est donc nécessaire de développer des approches alternatives, évolutives et abordables pour réduire la charge globale de morbidité.
« Notre motivation pour cette étude était double », a déclaré l’auteur co-correspondant Jian Xu, Ph.D., Département de pathologie de St. Jude et Centre d’excellence pour les études sur la leucémie. « Premièrement, pour découvrir comment l’édition du génome CRISPR inactive efficacement BCL11A pour la réactivation de l’hémoglobine fœtale. Et deuxièmement, pour identifier des stratégies thérapeutiques plus rentables et plus accessibles. »

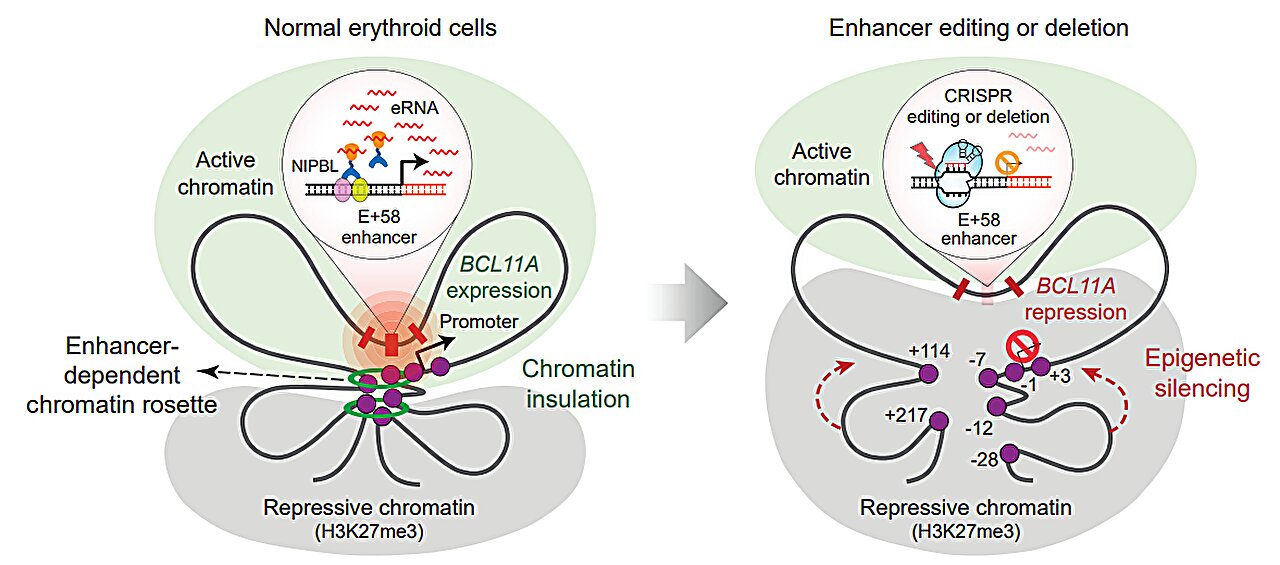
La thérapie génique brise la structure tridimensionnelle de l’ADN
L’équipe a étudié comment BCL11A est régulé au cours de l’hématopoïèse (croissance des cellules sanguines) et a identifié un mécanisme de contrôle clé : la région amplificatrice ciblée par la thérapie génique se repliait en une structure tridimensionnelle. « Nous avons découvert que cet activateur forme une structure de ‘rosette’ de chromatine, établissant de multiples contacts avec des éléments régulateurs critiques du gène », a expliqué Xu. « Cela garantit une expression de haut niveau de BCL11A et empêche son inactivation dans les précurseurs des globules rouges. »
Lorsque CRISPR-Cas9 effectue une cassure de l’ADN dans cet activateur dans le cadre de la thérapie, il perturbe la structure de la rosette de chromatine. Sans cette structure, d’autres protéines répressives peuvent pénétrer et faire taire le gène BCL11A.
De plus, les chercheurs ont découvert que la formation de cette structure complexe nécessite un type spécial d’ARN appelé ARN « amplificateur ». Ils ont testé si le ciblage de cet ARN amplificateur avec des oligonucléotides antisens, un type de thérapie plus rentable qui ne modifie pas le génome, pourrait produire le même bénéfice thérapeutique.
« En administrant des oligonucléotides antisens aux précurseurs des globules rouges normaux et falciformes, nous avons découvert que nous pouvons dégrader sélectivement l’ARN activateur, provoquant l’inactivation du BCL11A et la réactivation de l’hémoglobine fœtale », a déclaré Xu. « Nous pensons que cela pourrait offrir une alternative plus abordable, plus accessible et plus évolutive aux thérapies géniques actuelles. »