
La dégénérescence rétinienne de la rétinite pigmentaire est causée par une famille de mutations héréditaires dans près de 100 gènes qui conduisent lentement à la cécité au fil des années ou des décennies.
L’un de ces gènes code pour l’enzyme DHDDS, une partie de la voie qui glycosylate les protéines dans des cellules plus élevées. La rétinite pigmentaire des mutations DHDDS est appelée RP59. Il s’agit d’une maladie génétique récessive, ce qui signifie que des mutations doivent être présentes sur les deux copies du gène DHDDS pour provoquer une maladie.
Pour mieux comprendre et potentiellement traiter RP59, Steven Pittler, Ph.D., et ses collègues de l’Université de l’Alabama à Birmingham ont créé de nouveaux modèles de souris avec des mutations dans le gène de la souris pour les DHDD.
Leur premier modèle, signalé en 2020 et 2023, était un mutant K42E / K42E. La terminologie K42E / K42E est une sténographie génétique, ce qui signifie que les deux copies du gène DHDDS de souris avaient une mutation à l’acide aminé numéro 42, et ces mutations ont remplacé l’acide aminé de lysine (K) avec de l’acide glutamique (E). L’allèle K42E / K42E est observé dans la maladie de RP59 humaine.
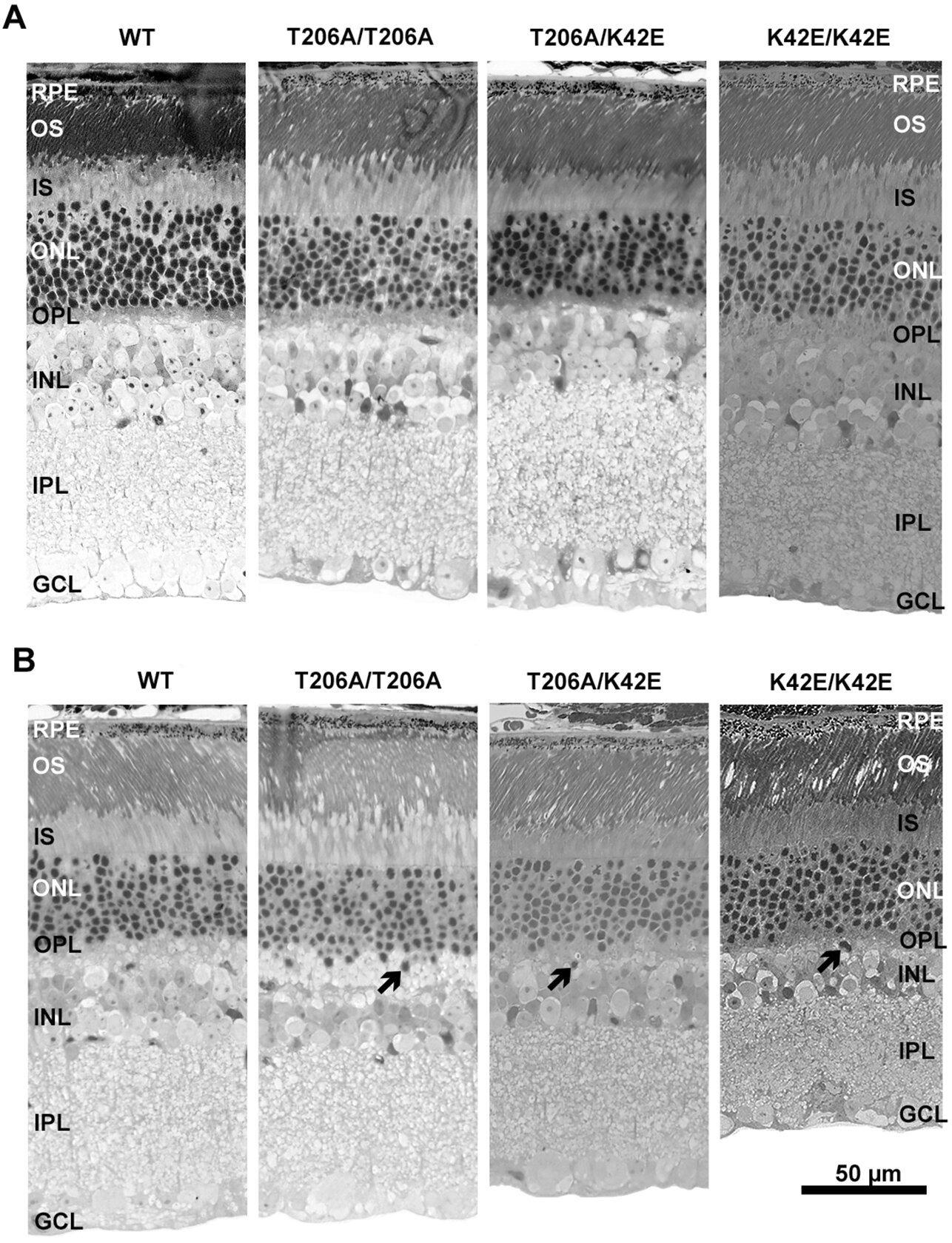
Les chercheurs UAB rapportent désormais deux autres modèles de souris: un mutant T206A / K42E et un mutant T206A / T206A. T206A signifie que l’acide aminé thréonine (T) 206 des DHDD a été remplacé par l’alanine (A). À 12 mois, les souris T206A / K42E et les souris T206A / T206A ont montré des changements dans la structure et la fonction rétiniennes similaires au modèle de souris K42E / K42E, selon l’étude publiée dans la revue Modèles et mécanismes de la maladie.
« Étant donné que T206A / K42E est l’une des variantes répandues rapportées chez les patients RP59, ces résultats nous rapprocheront de la compréhension du mécanisme sous-jacent à cette maladie », a déclaré Pittler, professeur au Département d’optométrie et de la science de la vision de l’UAB.
 Images représentatives de sections de tissu rétinien immunomarqué obtenues à partir de souris WT (A), K42E / K42E (B), T206A / T206A (C) et T206A / K42E (D). Crédit: Modèles et mécanismes de la maladie (2025). Doi: 10.1242 / dmm.052243")
Ces résultats indiquent que l’allèle DHDDS T206A, comme l’allèle K42E, provoque une maladie rétinienne, probablement par le biais d’un mécanisme pathobiologique commun, et nous proposons que la base physiologique de la dysfonction rétinienne dans RP59 implique une dégénération bipolaire et amacrine bipolaire.
Les cellules bipolaires sont des intermédiaires dans les couches de la rétine des cellules de rassemblement de lumière à l’arrière de la rétine au nerf optique, qui envoie ensuite ces informations au cortex visuel du cerveau.
Une constatation selon laquelle T206A / T206A provoque une maladie similaire aux modèles T206A / K42E et K42E / K42E montre que l’allèle T206A lui-même est pathogène, dit Pittler, même si l’allèle T206A / T206A n’a pas été vu chez l’homme.
Chez l’homme, l’allèle T206A / K42E provoque le même phénotype mais est moins robuste que K42E / K42E.
Les changements dans la structure et la fonction rétiniennes chez les souris T206A / T206A et T206A / K42E, comme celles rapportées plus tôt pour les souris K42E / K42E, ont été une réduction de l’épaisseur de la couche nucléaire interne et de la réduction des densités de cellules bipolaires et amacrines. L’électrorétinographie a révélé une réduction des ondes B, mais une réduction de l’épargne des amplitudes des ondes A. L’électrorétinographie utilise une électrode à la surface de l’œil pour mesurer les réponses électriques des neurones rétiniens à un éclair de lumière et déterminer quelles couches de neurones rétiniennes sont défectueuses.
La première réponse électrique, l’onde A, reflète la santé des cellules photorécepteurs dans la rétine externe qui détectent les photons. La deuxième réponse, la B-Wave, reflète la santé des couches intérieures de la rétine, qui se trouvent en aval des cellules photoréceptrices.