
Les scientifiques des National Institutes of Health (NIH) ont réussi à réduire la gravité des maladies de Tay-Sachs (lots) dans les cultures de cellules humaines et un modèle de souris en utilisant un nouveau traitement d’édition génétique.
Les lots sont une forme rare de maladie de Tay-Sachs, avec des signes et des symptômes tels que la faiblesse musculaire, la perte de coordination, les spasmes musculaires et parfois la perte de fonction mentale commençant à la fin de l’enfance à l’âge adulte. Des troubles similaires pour lesquels cette percée a des implications comprennent la gangliosidose de GM1, la maladie de Sandhoff, la maladie de Niemann-Pick, la maladie de Krabbe et la maladie de Gaucher.
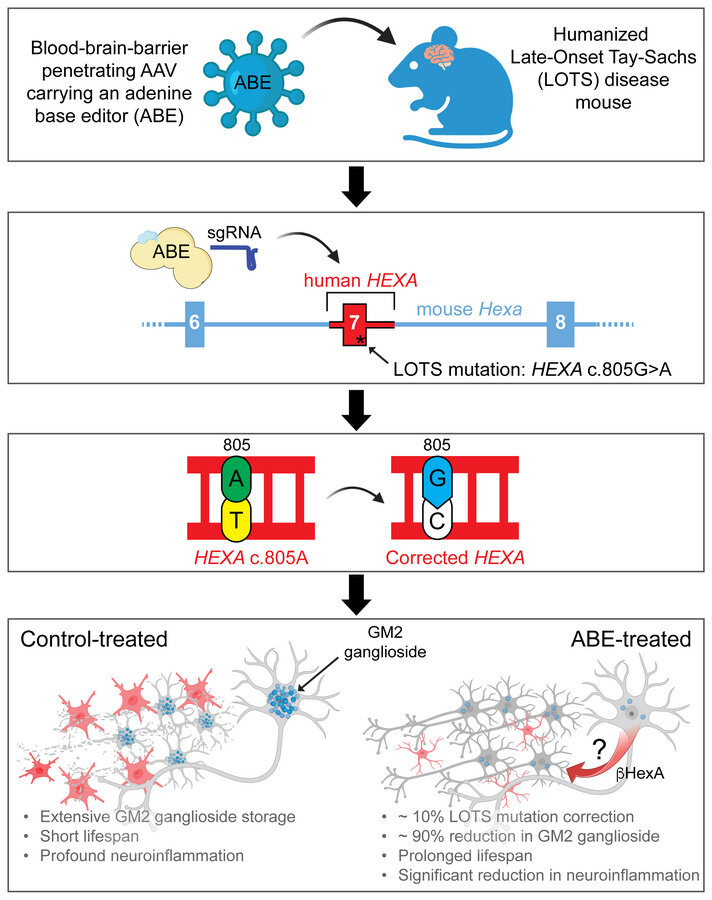
Les lots sont un trouble génétique provoqué par une mutation du gène hexa qui provoque une carence d’une enzyme qui est essentielle pour décomposer une substance graisseuse dans le cerveau, connu sous le nom de ganglioside GM2. L’accumulation de cette substance graisseuse endommage les cellules nerveuses dans le cerveau et la moelle épinière. La quantité d’enzyme encore produite par le corps affecte la gravité de la maladie et l’âge d’apparition.
En déploiement de la correction du gène Hexa, les scientifiques ont pu augmenter l’activité de l’enzyme, appelée bêta-hexosaminidase A, retarder l’apparition des symptômes et prolonger considérablement la durée de vie dans le modèle de la souris. La recherche est publiée dans le Journal of Clinical Investigation.
« Avec beaucoup de choses, une légère correction ira très loin. Ce montage peut avoir seulement besoin d’augmenter l’activité enzymatique d’environ 10% pour empêcher les symptômes de s’aggraver et d’améliorer leur qualité de vie », a déclaré l’auteur du journal, le Dr Richard Proia de l’Institut national du diabète et des maladies digestives et rénales. « Nous avons compris que l’ouverture de la porte à une activité enzymatique accrue est possible, nous devons maintenant comprendre comment le faire chez une personne. »
Les scientifiques pensent que ce niveau de travail préclinique a jeté les bases sur lesquelles construire vers les tests chez les participants humains. Les chercheurs estiment que les lots affectent environ 500 personnes dans le monde et ont actuellement environ 25 participants à une étude en cours au NIH Clinical Center de Bethesda, Maryland. Les cellules humaines utilisées dans cette étude ont été données par l’un des participants, qui est unique car ils ont deux copies du gène muté.
« Je n’ai jamais rencontré un participant à l’étude qui était si impatient et enthousiaste à faire partie du processus scientifique », a déclaré l’auteur de l’étude, le Dr Cynthia Tifft du National Human Genome Research Institute du NIH. « C’est inspirant de travailler avec quelqu’un qui reste positif et engagé pendant que ce trouble les prive de contrôle sur leur corps. Je suis motivé tous les jours en sachant que le travail que nous faisons est important. »
Bien que la percée actuelle ne soit pas encore un traitement ou un traitement viable, les chercheurs pensent qu’ils sont sur la voie d’un traitement potentiel. Les études futures étudieront les meilleures façons de fournir l’édition génétique au système nerveux central et au cerveau.
De nombreuses autres études d’édition génétique ont utilisé une méthode connue sous le nom de virus adéno-associé (AAV) pour fournir des modifications d’ADN aux cellules ciblées. Un AAV est un virus non enveloppé qui peut être conçu en tant que véhicule de livraison mais fait face à des problèmes chez de nombreux adultes qui ont peut-être déjà développé des anticorps contre certaines des particules de virus courantes utilisées pour le mécanisme d’administration.
Un autre obstacle consiste à concevoir une méthode de livraison qui peut traverser la barrière hémato-encéphalique, une zone où les vecteurs AAV peuvent avoir besoin d’amélioration.
Les chercheurs ont spécifiquement ciblé des lots pour cette recherche car d’autres formes de maladie de Tay-Sachs se produisent plus soudainement. La forme infantile du trouble est généralement diagnostiquée dans les 3 à 6 premiers mois de vie et est mortelle à l’âge de 4 à 5 ans. Les personnes avec beaucoup ont environ 4% à 6% de bêta-hexosaminidase A niveau d’activité enzymatique, tandis que les nourrissons n’ont aucune activité dans l’enzyme, rendant ainsi plus rapidement l’accumulation de ganglioside GM2 dommageable. Les enfants diagnostiqués avec la forme juvénile de la maladie meurent souvent à l’adolescence.
Les mutations du gène hexa qui provoquent la maladie de Tay-Sachs sont connues pour être trouvées plus souvent dans certaines populations, notamment les communautés juives d’Europe orientale et centrale (juifs ashkénazes), certaines communautés canadiennes françaises en Québec, la communauté cajun de la Louisiane et l’ancienne communauté Amish de l’Ordre en Pennsylvanie. Aux États-Unis, les femmes enceintes et leurs partenaires reçoivent souvent un test sanguin pour identifier les porteurs du changement de gène Hexa qui provoque une maladie de Tay-Sachs.