
En 1962, un résident en neurologie pédiatrique du Neurological Institute de Columbia et ses collègues du Collège des médecins et chirurgiens ont publié un article détaillé dans Pédiatrie décrivant un nouveau syndrome de neurodégénérescence chez les nourrissons de sexe masculin.
Le résident, John Menkes, MD, avait vu le cas index quelques années plus tôt à l’hôpital pour bébés (maintenant l’hôpital pour enfants Morgan Stanley du NewYork-Presbyterian/Columbia University Irving Medical Center). Le garçon avait atteint les premières étapes de son développement, telles que le contrôle de la tête et le sourire, à l’âge de six semaines. Mais ensuite, l’enfant a brusquement régressé. Il a perdu du poids et du contrôle musculaire et a rapidement développé des convulsions fréquentes. Rien de ce que Menkes a fait n’a semblé aider et l’enfant est décédé à l’âge de 18 mois.
Après avoir examiné les dossiers de quatre autres nourrissons de sexe masculin de la même famille de New York qui étaient arrivés à l’hôpital pour bébés avec des symptômes similaires, Menkes en a déduit qu’il s’agissait d’une maladie génétique récessive liée à l’X. Une décennie plus tard, la cause a été identifiée comme une carence en cuivre, une découverte qui a révélé le besoin crucial du cerveau humain en ce métal trace. Une étude plus approfondie révélerait également la rareté de la maladie, qui touche environ 1 naissance vivante de sexe masculin sur 35 000.
« C’est une maladie tragique », déclare Stephen G. Kaler, MD, professeur de pédiatrie récemment recruté au Collège des médecins et chirurgiens de Vagelos, qui dirige la clinique de la maladie de Columbia Menkes. « Les nourrissons atteints qui naissent en bonne santé tombent malades en quelques semaines, avec des convulsions, un retard de croissance et des retards de développement. Sans traitement, il y a une spirale inexorable vers le bas et la mort survient souvent avant l’âge de deux ans. C’est évidemment très dur pour les parents. »
À la fin des années 1980, Kaler était postdoctorant dans un laboratoire du NIH étudiant le métabolisme du cuivre humain lorsqu’il a été confronté pour la première fois à la maladie de Menkes, comme on l’appelait désormais.
« Il y avait plusieurs troubles héréditaires du métabolisme du cuivre connus à cette époque », explique Kaler. « La maladie de Menkes était celle qui semblait nécessiter le plus d’attention, tant du point de vue de la science fondamentale que du traitement, c’est pour cette raison que j’y ai été attirée. »
Trois décennies plus tard, les thérapies développées grâce aux recherches de Kaler pourraient bientôt changer les perspectives des enfants nés avec Menkes. L’un d’entre eux, un médicament qui augmente considérablement la survie des patients atteints de Menkes, appelé histidinate de cuivre (CuHis), est actuellement en cours d’examen pour l’approbation d’un nouveau médicament par la FDA. Et une thérapie génique virale développée par le laboratoire de Kaler, combinée à CuHis, pourrait offrir aux enfants des avantages encore plus importants – et éventuellement un avenir à long terme.
Un premier départ avec les injections de cuivre
La première tentative de Kaler au début des années 1990 pour traiter Menkes a commencé avec une supplémentation en cuivre. Une équipe au Canada avait expérimenté l’administration de CuHis à deux enfants atteints de la maladie de Menkes pour augmenter les niveaux de cuivre, et Kaler a lancé un protocole au centre clinique des NIH.
« C’était une opportunité unique, car au NIH, je pouvais traiter des patients de tout le pays et du monde entier atteints de cette maladie rare et évaluer formellement son efficacité dans le cadre d’essais cliniques. »
Les premiers résultats ont été mitigés, dit Kaler. Il a appris que les enfants présentant des variantes plus légères du gène de transport du cuivre de Menkes réagissaient mieux à la supplémentation en cuivre, tout comme les enfants traités peu après la naissance.
Une expérience plus longue avec un plus grand nombre de patients a identifié des avantages distinctifs en termes de survie et de résultats cliniques, même avec des variantes de perte de fonction sévère, utilisant une formulation CuHis produite au NIH et plus tard par Cyprium Therapeutics et Sentynl Therapeutics, partenaires biopharmaceutiques.
Parmi les patients traités peu après la naissance, la survie médiane est passée à 15 ans ; certains participants sont même allés à l’université, ont rejoint le marché du travail et mènent une vie presque normale. Les enfants traités plus tard, qui avaient déjà développé des symptômes, ont également montré un bénéfice en termes de survie, vivant en moyenne jusqu’à l’âge de cinq ans.
« Les CuHis ont donné aux parents plus de temps avec leurs enfants, et je sais qu’ils ont profondément apprécié cela », a déclaré Kaler. « Mais il semblait que pour traiter la maladie de manière plus complète, nous avions également besoin d’une stratégie de remplacement génétique. »
Les injections introduisent du cuivre dans la circulation sanguine, explique Kaler, mais comme des erreurs dans le gène Menkes altèrent le transport du cuivre, de nombreuses cellules, y compris les neurones du cerveau, ne reçoivent pas suffisamment de cet élément.
Une thérapie génique associée à une supplémentation en cuivre résout ces deux problèmes.
Succès de la thérapie génique dans les études animales
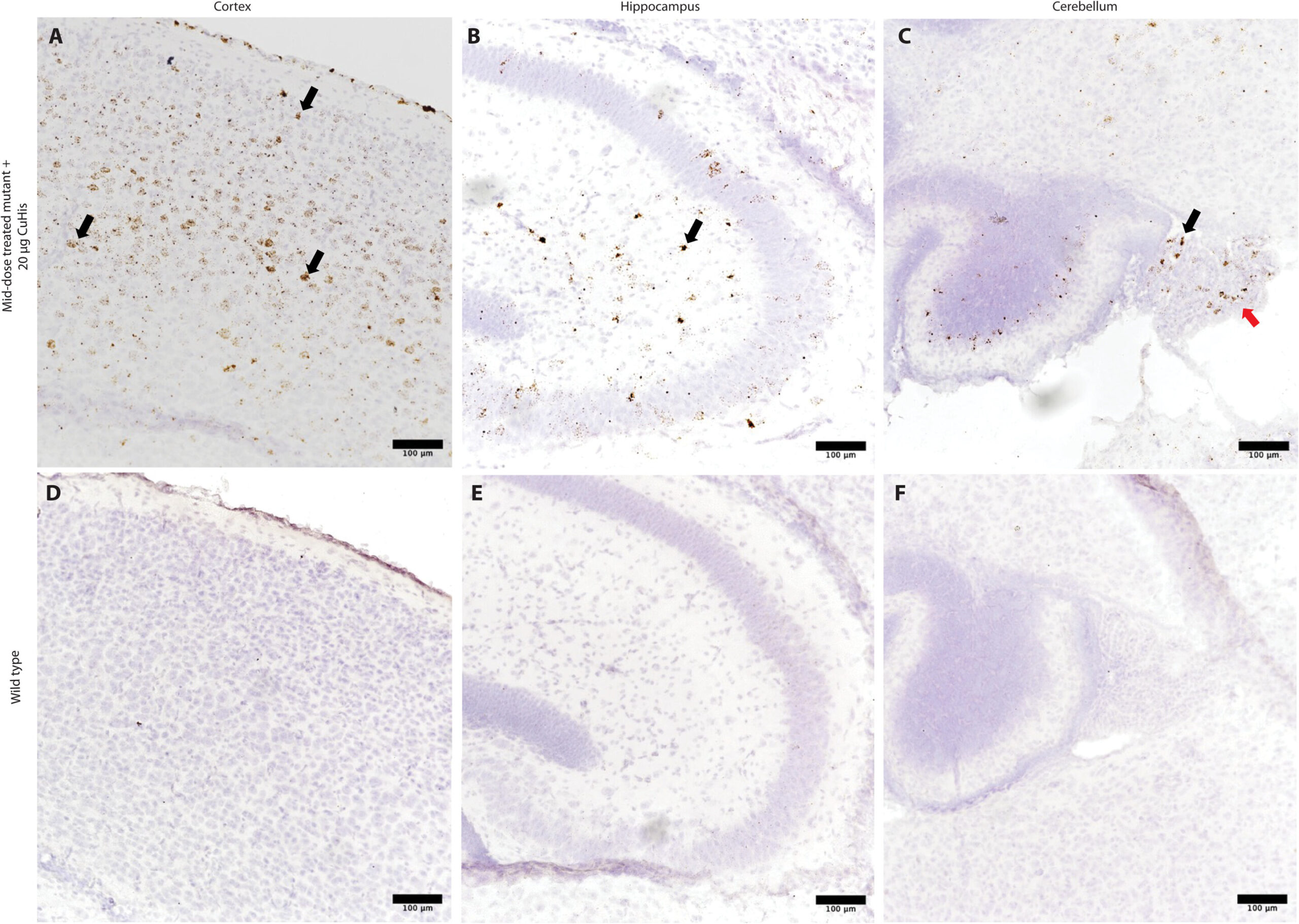
Avant de rejoindre Columbia en août 2024, Kaler a quitté le NIH pour travailler avec des pionniers de la thérapie génique virale au Nationwide Children’s Hospital et à l’Ohio State University à Columbus, Ohio, pour créer une thérapie génique Menkes. La thérapie, injectée dans la circulation sanguine, délivre un gène transporteur de cuivre fonctionnel au cerveau.
Les premiers tests précliniques de la dernière conception de Kaler ont été publiés en août 2025 dans Avancées scientifiques et montrent des résultats remarquables lorsqu’ils sont associés à des injections de CuHis : une dose unique de thérapie génique atteint le cerveau et prévient la maladie chez près de 100 % des souris présentant un défaut du gène Menkes.
« Nous devons effectuer des tests supplémentaires sur les animaux pour confirmer la sécurité de cette thérapie génique afin qu’elle puisse être utilisée chez des sujets humains, et je pense que nous sommes probablement dans un an ou deux avant de commencer un essai clinique du traitement combiné », a déclaré Kaler.
« Nous espérons qu’avec les progrès du dépistage néonatal permettant une détection précoce, le traitement de la maladie de Menkes avec CuHis et la thérapie génique modifieront l’histoire naturelle de cette maladie », a déclaré Kaler. « Cela bouclerait une boucle dans une histoire pour laquelle le centre médical Irving du New York Presbyterian/Columbia University et le NINDS ont joué un rôle si important. »
« Il a été dit à propos de certains efforts qu’il n’y avait pas de solution miracle et que les progrès devaient être mesurés en décennies. La perte d’un jeune enfant est l’une des plus grandes tragédies de la vie, et les progrès récents et les approbations potentielles de la FDA pour ces traitements sont satisfaisants. Il y a encore du travail à faire, bien sûr, mais au moins maintenant nous avons une voie à suivre rationnelle et pouvons imaginer un jour où la vie des enfants et de leurs parents ne sera plus assombrie par cette maladie difficile. «