
Une équipe dirigée par l’Institut de Neurociències de l’UAB (INc-UAB) a identifié pour la première fois le mécanisme à l’origine d’un type d’épilepsie potentiellement mortel, un symptôme des maladies mitochondriales. La découverte, basée sur des modèles murins, remet en question l’idée traditionnelle selon laquelle ces troubles sont causés par un déficit énergétique généralisé et révèle plutôt un dysfonctionnement précis de circuits cérébraux spécifiques. La recherche ouvre la voie à des thérapies contre cette forme mortelle d’épilepsie basées sur la normalisation de la fonction des neurones du noyau sous-thalamique.
Les maladies mitochondriales sont un groupe de troubles très graves provoqués par des défauts des mitochondries, la machinerie chargée de produire l’énergie cellulaire. Ils touchent environ 1 naissance sur 4 300. En raison de leur faible incidence, elles sont considérées comme des maladies rares et peu de ressources sont donc réservées à leur recherche.
Toutes les cellules du corps ne sont pas également vulnérables au dysfonctionnement mitochondrial : celles qui ont des besoins énergétiques plus élevés ont tendance à dégénérer plus rapidement. Étant l’un des organes les plus consommateurs d’énergie, le cerveau est l’un des premiers à être touché et dans une plus large mesure. En ce sens, l’un des symptômes les plus courants et particulièrement graves est une forme d’épilepsie qui ne répond pas aux traitements conventionnels.
Maintenant, dans une nouvelle étude publiée dans le Journal d’investigation cliniqueune équipe de recherche dirigée par l’Institut de Neurociències de l’Universitat Autònoma de Barcelona a pu approfondir ses recherches sur ce type d’épilepsie. En utilisant des modèles murins atteints du syndrome de Leigh, l’une des maladies mitochondriales les plus fréquentes, les chercheurs ont pu étudier des circuits cérébraux spécifiques et les altérations neuronales qui les provoquent.
Les crises d’épilepsie impliquant une activation neuronale excessive, l’équipe a travaillé sur les neurones inhibiteurs, appelés neurones GABAergiques (qui libèrent le neurotransmetteur inhibiteur GABA), avec l’hypothèse que ces neurones ne fonctionnaient peut-être pas correctement. Les scientifiques soupçonnaient que le problème pourrait provenir d’une perte du contrôle naturel des inhibiteurs du cerveau.
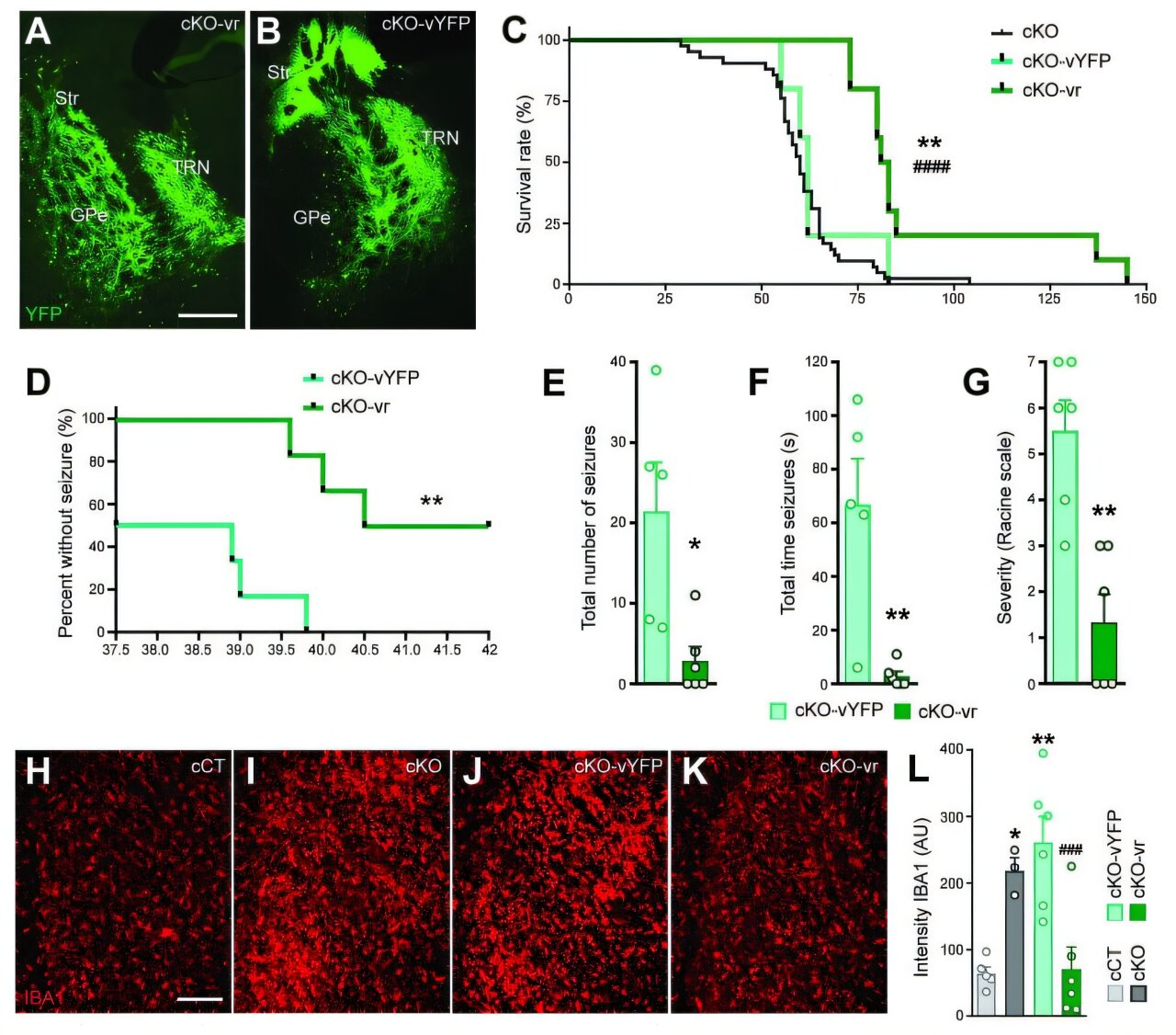
« Nous avons émis l’hypothèse que le dysfonctionnement mitochondrial des neurones GABAergiques reliant deux régions clés du cerveau, le globus pallidus externe (GPe) et le noyau sous-thalamique (STN), pourrait être à l’origine de l’hyperexcitabilité observée dans ce circuit », explique Laura Sánchez-Benito, chercheuse à l’INc-UAB et première auteure de l’étude. étude.
L’équipe a utilisé deux modèles différents de souris atteintes du syndrome de Leigh pour tester l’hypothèse : le modèle KO, dans lequel la fonction mitochondriale est détériorée dans tout le corps et la plupart des symptômes de la maladie sont reproduits ; et le modèle cKO, dans lequel le dysfonctionnement mitochondrial n’affecte que les neurones GABAergiques du GPe, permettant d’isoler les effets de ce défaut localisé.
Grâce à des techniques avancées d’imagerie, électrophysiologiques et génétiques, l’équipe a découvert que les neurones GPe GABAergiques sont particulièrement vulnérables au dysfonctionnement mitochondrial, qui conduit à leur dégénérescence généralisée. Puisque ces neurones sont chargés de maintenir sous contrôle l’activité du STN, leur perte rend le noyau hyperactif, ce qui produit des crises d’épilepsie récurrentes chez les animaux.
En revanche, lorsque l’équipe a restauré les fonctions mitochondriales du GPe, les crises ont presque disparu. Les souris ont vécu plus longtemps et ont montré une amélioration notable de leurs fonctions neurologiques. « Nous avons été surpris de voir comment la perte d’une seule population de neurones inhibiteurs pouvait se propager à travers le réseau et déclencher des crises d’épilepsie », explique Sánchez-Benito.
« Notre étude démontre que ce n’est pas l’ensemble du cerveau qui propulse la crise, mais plutôt que restaurer la fonction de ces neurones spécifiques suffit à supprimer drastiquement cette forme mortelle d’épilepsie », affirme le professeur Albert Quintana, coordinateur de l’étude et chercheur à l’INc-UAB et au Département de biologie cellulaire, physiologie et immunologie de l’UAB. « De plus, comme l’inactivation du STN par stimulation cérébrale profonde est déjà une thérapie utilisée, cette option pourrait être envisagée pour les patients atteints du syndrome de Leigh », conclut-il.
L’étude, menée en collaboration avec des chercheurs de l’Institut de Neurosciences d’Alacant, de l’Institut de Recherche de Vall d’Hebron (VHIR) et du Département de Biochimie et Biologie Moléculaire de l’UAB, ouvre de nouveaux horizons aux patients atteints de maladies mitochondriales qui n’ont actuellement aucune option thérapeutique pour traiter leurs crises.